
Genomic signatures of local selection in Atlantic salmon
Understanding how populations adapt to their local environment both improves our understanding of how biodiversity evolves, and guides our management of threatened or exploited taxa. With Craig Primmer at the University of Turku, I am using dense genomic datasets to identify loci potentially underlying local adaptation in Atlantic salmon (Salmo salar). My main study system is the large sub-Arctic Teno River salmon stock, which includes many genetically distinct sub-populations. Comparing juvenile fish from different locations, I found signals suggesting differential local selection around genes associated with sexual maturation, energy homeostasis and immune defence. These included the large-effect age-at-maturity gene vgll3 , the well-known obesity gene mc4r, and major histocompatibility complex II. Most strikingly, I confirmed a region on Chromosome 9 that is extremely differentiated among Teno sub-populations and is also a candidate for local selection over the global range of Atlantic salmon. This region overlaps with a haplotype that underlies spawning ecotype in sockeye salmon (Oncorhynchus nerka), suggesting that the same gene may be the selective target in both cases. The phenotypic effect of this region in Atlantic salmon is unknown, although allelic variation covaries with upstream catchment area and timing of the return spawning migration. My ongoing follow-up work examining Atlantic salmon populations in north-western Russia, and comparing results from similar studies, indicates that many of the same loci could be adaptively important in Atlantic salmon at regional and continental scales, as well as in other salmonid species. This research is part of the Integrative Science for Adaptive Co-Management in the Arctic project.

Factors influencing hybridization between wild salmon and aquaculture escapees
Wild Atlantic salmon are threatened by introgressive hybridization with domestic salmon that have escaped from fish farms. While some wild populations have shown genetic changes as a result of this interbreeding, other populations have retained their genetic integrity despite receiving similar numbers of escapees. It is unknown what makes an Atlantic salmon population vulnerable to such 'genetic invasion'. I am addressing this question by studying wild-escapee hybridization in the Teno River using a 45 year scale archive. I have identified a set of SNP markers that can collectively discriminate different classes of aquaculture-wild hybrids. Currently, I am using these to investigate hybridization dynamics over space and time with the aim of identifying environmental and demograpic factors that influence these dynamics.

Regulatory architecture of gene expression variation in the threespine stickleback
Adaption to novel environments ofteninvolves changes in regions of the genome that regulate gene expression, rather than in the coding regions of genes themselves. The threespine stickleback (Gasterosteus aculeatus) is an important model for local adaptation, yet the regulatory architecture of gene expression variation is not well characterized in this species. Working with Erica Leder and Craig Primmer at the University of Turku I combined gene expression and RAD-tag datasets for sib/half sib stickleback families and used a QTL mapping approach to identify regions of the genome underlying expression variation in over 10,000 genes. Results suggested the presence of several QTL ‘hotspots’ that influenced the expression of multiple, distant genes. However, these ‘hotspots’ did not overlap with regions of the genome known to be involved in stickleback adaptive divergence.

Conservation genetics of cutthroat trout
The interior cutthroat trout (O. clarki subsp.) of western North America have declined to a fraction of their native range as a result of habitat loss and hybridization with introduced rainbow trout (O. mykiss). In collaboration with David Cowley at New Mexico State University, Jessica Metcalf and Andrew Martin at the University of Colorado, John Carlos Garza at the Southwest Fisheries Science Center and Mary Peacock at the University of Nevada, I used population genetic approaches to guide cutthroat trout management. My work showed that population fragmentation has had important genetic consequences; that a signature of evolutionary independence can be retained between drainages despite large scale transplants of fish between them; but that such historic transplants could also result in populations being mis-identified and containing unexpected genetic contributions from other cutthroat subspecies. I also found that levels of non-native introgression estimated using existing non-diagnostic markers such as microsatellites varied with the statistical method and pure reference sample used. This conflicted with recommendations placing cutthroat trout populations into different management categories based on threshold introgression levels. To address this problem, I subsequently developed a large set of SNP markers diagnostic between rainbow trout and all cutthroat trout subspecies.
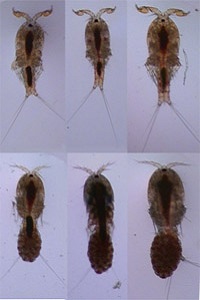
Consequences of inter-population hybridization in Tigriopus californicus
Although introgressive hybridization with introduced taxa is a threat to many species of conservation concern, the consequences of this hybridization are difficult to predict. Examining the outcomes of inter-population hybridization in the laboratory can both inform the management of such threatened taxa, and shed light on genomic interactions that contribute to reproductive barriers between species. I undertook this work with Suzanne Edmands at the University of Southern California, using the intertidal copepod Tigriopus californicus - an important model for outbreeding depression. Genotypic deviations from Hardy-Weinberg equilibrium in controlled crosses between populations suggested that hybrid fitness was influenced both by nuclear-nuclear epistatic interactions and by the presence of deleterious recessive alleles in the parental populations. Genomic outcomes in long term, freely mating, hybrid swarms partly reflected random factors: variation among replicates in the initial contribution of pure parents, and genetic drift. Thus, the outcome of a hybridization event in nature may not be predictable. However, deterministic factors were also important: where pure parents exhibited differential fitness in the lab, genetic material from the fitter population rapidly dominated a swarm. Thus, an invasive species that is more fit in the local environment may rapidly cause 'genetic swamping' of the native population. Different parts of the genome exhibited differential patterns of introgression, which could be strikingly similar across replicate swarms and reflected the genotypic distortion previously observed in controlled crosses. This suggested that deleterious genotypic combinations were removed by selection over time - correspondingly, hybrid swarms showed no long-term decrease in fitness compared to their pure parents. Thus, even with reduced fitness in initial generations, populations may be able to recover from the deleterious effects of non-native introgression.
